Fat a culprit in fibrotic lung damage
Pulmonary fibrosis has no cure. It's caused by scarring that seems to feed on itself, with the tougher, less elastic tissue replacing the ever moving and stretching lung, making it increasingly difficult for patients to breathe. Researchers debate whether the lung tissue is directly damaged, or whether immune cells initiate the scarring process - an important distinction when trying to find new ways to battle the disease. Now research shows that both processes may be important, and suggest a new direction for developing novel therapies. The work will publish online November 20th in the American Journal of Respiratory Cell and Molecular Biology.
"By changing our focus, not just to lung cells or immune cells, but to how these cells might be communicating, we may find new opportunities for treating pulmonary fibrosis," says Ross Summer, M.D., Associate Professor of in the department of Pulmonary and Critical Care Medicine at Thomas Jefferson University, who studies the disease and regularly treats patient with this illness.
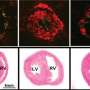
"In the advanced stages of the disease, there's not a lot we can do for patients," says Dr. Summer. Some existing therapies alleviate symptoms, but none reverse or stop disease progression. Many patients live only three to five years after diagnosis, according to the American Lung Association and the only effective treatment is lung transplant. The team led by Dr. Summer and first author Freddy Romero, Ph.D., looked at a mouse model of lung fibrosis initiated by a chemical known to cause the disease. Researchers noticed that lipids (AKA fat), accumulated within the airspaces of the lung where oxygen is absorbed. Although lipids are normally secreted there to help keep the cells lining the lungs lubricated and properly inflated, these were excessive levels of fat.
The researchers showed that in response to stress, the cells producing the lubricant dump their lipid stores into the lungs and fail to mop up the excess. The excess lipids react with oxygen to create a form of fat that acts as an inflammatory signal; in some ways this response is similar to the events that initiate atherosclerosis, or plaque formation in blood vessels. In the lungs, Dr. Summer's laboratory showed that immune cells called macrophages, which normally survey the lung for debris, infection, or dying cells begin gobbling up the excess fat in the lungs. Loaded with this oxidized fat, the macrophages turned on a program that acts to help heal the wounded tissue, but as a consequence to this adaptive response leads to the development of fibrotic lung disease.
"Both the initial damage to the cells lining the airway of the lung and the inflammation are important," says Dr. Romero, "but the thing that drives the damage is the unregulated excess lipids in the distal airspaces." When the researchers put oxidized lipids into the lungs of mice that had not been exposed to any lung-damaging chemicals, the mice also developed fibrosis, showing that the oxidized fat alone was enough to cause the disease.
"These results show, for the first time, that a break-down of normal lipid handling may be behind this lung disease," says Dr. Summer "If we prove that the same process holds true in humans, it suggests that we could prevent or mitigate the disease by simply clearing out the excess oxidized lipids from lungs."
To this end, the researchers tested whether treating mice with an agent called GM-CSF that reduces lipid secretion and facilitates lipid removal in the lungs, could minimize lung fibrosis. Indeed, this agent reduced the scarring in the lungs by over 50 percent based on the levels of lung collagen, a marker of newly forming scar tissue. In addition, the researchers examined human cells in the lab and saw that oxidized fat also promoted a fibrotic response.
Future work will focus on exploring whether the same results hold true in humans.
More information: F. Romero et al., "A pneumocyte-macrophage paracrine lipid axis drives the lung toward fibrosis," Am J Respir Cell Mol Biol, DOI: 10.1165/rcmb.2014-0343OC , 2014.







.jpg)