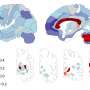
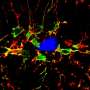
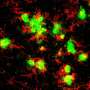
Protein that regulates brain cell connections could be new target for treating Alzheimer's disease

In experiments with a protein called Ephexin5 that appears to be elevated in the brain cells of Alzheimer's disease patients and mouse models of the disease, Johns Hopkins researchers say removing it prevents animals from developing Alzheimer's characteristic memory losses. In a report on the studies, published online March 27 in The Journal of Clinical Investigation, the researchers say the findings could eventually advance development of drugs that target Ephexin5 to prevent or treat symptoms of the disorder.
"Ephexin5 is a tantalizing pharmaceutical target because in otherwise healthy adults, there's very little present in the brain," says Gabrielle L. Sell, a graduate student at the Johns Hopkins University School of Medicine. "That means shutting off Ephexin5 should carry very few side effects," adds Sell, who works with Seth S. Margolis, Ph.D., assistant professor of biological chemistry and neuroscience.
Their work with Ephexin5 grew out of a paradox about one of Alzheimer's disease's defining features, the development of thick plaques in the brain composed of a protein called amyloid beta. Stemming the production of this protein is currently the major focus of efforts to develop new Alzheimer's treatments, explain Sell and Margolis, but it isn't the amount of amyloid beta in patients' brains that correlates best with the severity of symptoms; rather, it's the loss of so-called excitatory synapses, a type of cellular structure forged between two brain cells.
Although it's not clear how amyloid beta and excitatory synapse loss are connected, researchers from the University of California, San Francisco, showed several years ago that Alzheimer's patients have decreased brain levels of a protein called EphB2. Margolis and his colleagues have focused on Ephexin5, a protein regulated by EphB2 and thought to be responsible for inhibiting the development of dendritic spines, small protrusions on the ends of nerve cells that are the location for most excitatory synapses.
Curious about whether Ephexin5 might also play an important role in Alzheimer's disease symptoms, Margolis, Sell and Thomas B. Schaffer, also a graduate student in Margolis' lab, first investigated whether this protein might be poorly regulated in Alzheimer's animal models and patients.
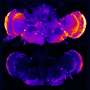
The researchers discovered that when they added amyloid beta to healthy mouse brain cells growing in petri dishes, these cells began overproducing Ephexin5. Additionally, when they injected the brains of healthy mice with amyloid beta, cells there also began overproducing Ephexin5—both clues that the protein that makes Alzheimer's characteristic plaques appears to trigger an increase in brain cells' production of Ephexin5 of between 1- and 2.5-fold.
When the researchers examined preserved brain tissues isolated from Alzheimer's patients during autopsies, they also found similarly high levels of Ephexin5. Additionally, they found elevated levels of Ephexin5 in mice genetically engineered to overproduce amyloid beta, and that show memory deficits similar to those with human Alzheimer's disease, further confirming that excess Ephexin5 is associated with this disease.
Armed with what they called this wealth of evidence that brain cells produce too much Ephexin5 when Alzheimer's disease linked to amyloid beta is present, the researchers then investigated whether reducing Ephexin5 might prevent Alzheimer's deficits.
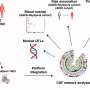
Using genetic engineering techniques that knocked out the gene that makes Ephexin5, the researchers developed mouse Alzheimer's disease models whose brain cells could not produce the protein. Although the animals still developed the characteristic Alzheimer's amyloid plaques, they didn't lose excitatory synapses, retaining the same number as healthy animals as they aged.
To see whether this retention of excitatory synapses in turn affected behavior related to memory tasks, the researchers trained healthy mice, mouse models of Alzheimer's and Alzheimer's models genetically engineered to lack Ephexin5 in two learning tasks: one that involved the ability to distinguish objects that had moved upon subsequent visits to the same chamber, and another that involved the ability to avoid chambers where they'd previously received a small electric shock.
While the typical Alzheimer's disease model mice appeared unable to remember the moved objects or the shocks, the Alzheimer's animals genetically engineered to be Ephexin5-free performed as well as healthy animals on the two tasks.
Margolis cautions that while the results all suggest removing Ephexin5 prevented Alzheimer's disease-associated impairments, they don't on their own provide a true test for the approach to treatment. That's because in people with Alzheimer's disease, the brain is exposed to amyloid beta for some time, probably decades, before any treatments might be administered.
To better reflect that human scenario, the researchers raised mouse models for Alzheimer's disease into adulthood—allowing their brains to be exposed to excess amyloid beta for weeks—before injecting their brains with a short piece of genetic material that shut down Ephexin5 production. These mice performed just as well on the memory tasks as the healthy mice and those genetically engineered to produce no Ephexin5.
Together, these results, say Margolis and Sell, suggest that too much Ephexin5 triggered by amyloid beta and reduced EphB2 signaling might be the reason why Alzheimer's disease patients gradually lose their excitatory synapses, leading to memory loss—and that shutting down Ephexin5 production could slow or halt the disease.
The team is currently investigating whether drugs currently in clinical trials for Alzheimer's disease could be exerting effects on Ephexin5 and how brain cells naturally regulate Ephexin5.
"This study gives us some hope that moving beyond efforts to interrupt amyloid beta pathways, and targeting pathways for synapse formation, will give us potent therapies for this devastating disease," Margolis says.
An estimated 5.1 million Americans have Alzheimer's disease. It's the most expensive disease in this country—care for patients in 2017 is estimated to cost $259 billion. More than 15 million Americans provide unpaid care for people with Alzheimer's and other dementias.