Potential treatment identified for bladder cancers that are ARID1A-deficient

Metastatic bladder cancer is generally incurable, so new therapies are an urgent need. Researchers at the University of Alabama at Birmingham now report a potential treatment for a quarter of bladder cancers.
Their discovery, published in the journal JCI Insight, comes from mechanistic insights of gene changes and altered signaling pathways that revealed therapeutically actionable targets and mechanisms of resistance.
The UAB research focused on mutations in the gene ARID1A, a key part of a chromatin-modifying complex in the cell nucleus. Chromatin is a mix of DNA and proteins that tightly packages the 5-foot-long chromosomal DNA inside each cell, and chromatin modifiers act to help turn on or turn off gene expression. ARID1A-inactivating mutations are seen in about 20 percent of bladder tumors
The foundations for this research were various previous studies in cancer. First, a histone methyltransferase called EZH2 is considered to be an oncogene that causes normal cells to become cancer cells because it is overexpressed in many aggressive cancers and is thought to drive growth of the cells as shown by the lead authors and others. EZH2 can silence genes, including those that act to suppress growth of a tumor, called tumor suppressors. Second, previous studies have shown that mutations in ARID1A sensitize cells to pharmacologic inhibition with the EZH2-specific small molecule GSK-126.
So the researchers, led by James "Jed" Ferguson III, M.D., Ph.D., and Sooryanarayana Varambally, Ph.D., hypothesized that bladder cancer cells with ARID1A mutations would show sensitivity to EZH2 inhibition, which could be utilized as a therapeutic target for patients with ARID1A-deficient bladder cancer. Ferguson is an assistant professor in the UAB Department of Urology, and Varambally is a professor in the UAB Department of Pathology Division of Molecular and Cellular Pathology.
Analysis of existing datasets by the researchers showed that up to 29 percent of bladder cancers have nonsense or truncating mutations in ARID1A, and these tumors also have high expression of EZH2. Furthermore, several existing bladder cancer cell lines have mutations in ARID1A, which the researchers call ARID1Amut, while other bladder cancer cell lines did not have ARID1A mutations.
Using these cell lines, the researchers showed that the ARID1Amut cancer cells—but not ARID1A wild-type cancer cells—were sensitive to EZH2 inhibition by GSK-126 in cell culture, as measured by viability and proliferation. In mouse xenografts, using both the bladder cancer cell lines and patient-derived bladder cancer cells, GSK-126 halted growth of cancer cells containing ARID1Amut but not those with wild-type ARID1A.
Ferguson and Varambally next hypothesized that ARID1A-deficient cells become sensitive to GSK-126 due to transcriptional upregulation of specific tumor suppressors that remain transcriptionally repressed in ARID1A wild-type cells. They compared gene expression of GSK-126-treated ARID1A-wild-type bladder cancer cell lines with GSK-126-treated ARID1A knockdown cell lines. The ARID1A knockdown cells have less ARID1A and show a sensitivity to GSK-126 inhibition similar to that of ARID1Amut cells.
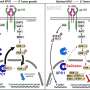
Out of about 70 differentially expressed genes, one was notable—the tumor suppressor PIKIP1 that acts by attenuating the cell-signaling pathway PI3K/AKT/mTOR. This pathway is important in regulating cell proliferation and growth in healthy cells; but in many cancers, it is overactive and causes unrestrained cell proliferation.
Healthy cells with wild-type ARID1A use the MAPK signaling pathway, but the researchers found that cells with ARID1Amut or ARID1A knockdowns use a different pathway. These cancer cells are dependent on PI3K signaling to activate the PI3K/AKT/mTOR pathway for survival, a signaling that is due to an upregulation of a relatively uncharacterized regulatory subunit of PI3K called PIK3R3.
Researchers confirmed this in clinical lysates of human bladder cancers, showing that tumors deficient in ARID1A protein had elevated levels of PIK3R3 and phosphoAKT, where phosphoAKT is the active form of AKT in the PI3K/AKT/mTOR pathway.
Of potential clinical importance, the researchers showed that ARID1A-deficient bladder cancer was sensitive to combination therapies with the EZH2 inhibitor GSK-126 and several inhibitors of PI3K, acting together in a synergistic manner.
"Thus, our studies suggest that bladder cancers with ARID1A mutations can be treated with inhibitors of EZH2 and/or PI3K," Ferguson said. Ferguson and Varambally note that ARID1A is frequently mutated across a wide variety of human cancers, including bladder, gastric, pancreatic and ovarian cancers. With FDA approved EZH2 inhibitors available, the lead authors discussed the possibility of targeting EZH2 in cancer in a previous review that appeared in the journal Cancer Research.
Further mechanistic studies by the researchers showed that the EZH2-inhibitor sensitivity in ARID1A-deficient bladder cancer cells is due to upregulation of PIK3IP1, a protein inhibitor of PI3K signaling. The researchers also showed, for the first time, that PIK3IP1 inhibits PI3K signaling by inducing proteasomal degradation of PIK3R3. In support of these findings, they showed that PIK3R3 upregulation is necessary and sufficient for PI3K/AKT pathway activation and increased bladder cancer cell proliferation. Similarly, they showed that PIK3IP1 upregulation was necessary and sufficient for GSK-126-mediated cell death in ARID1A-deficient bladder cancer cells.
More information: Hasibur Rehman et al, ARID1A-deficient bladder cancer is dependent on PI3K signaling and sensitive to EZH2 and PI3K inhibitors, JCI Insight (2022). DOI: 10.1172/jci.insight.155899
Marie-Lisa Eich et al, EZH2-Targeted Therapies in Cancer: Hype or a Reality, Cancer Research (2020). DOI: 10.1158/0008-5472.CAN-20-2147