Driver of cystic fibrosis lung inflammation yields target for treatment
. DOI: 10.1016/j.celrep.2022.111797")
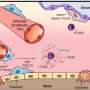
Yale researchers have identified a possible driver of the persistent inflammation that causes irreversible lung damage in patients with cystic fibrosis, a genetic disorder that impairs breathing and digestion.
In a new study, they uncover how a type of white blood cell called a monocyte kicks off a molecular chain of events that leads to sustained inflammation in the lungs and lung tissue damage. They also found that a drug that targets the monocytes was able to slow the progression of tissue damage in a mouse model of cystic fibrosis, suggesting it could be an effective treatment for cystic fibrosis in the future.
The findings were published Dec. 13 in Cell Reports.
Cystic fibrosis is a genetic disease that affects different organs in the body. In the lungs, the disease causes mucus buildup that traps bacteria and makes patients more susceptible to infection. Over time, as symptoms worsen, infections often become chronic throughout the remainder of the patients' lives.
Chronic inflammation is another complication of cystic fibrosis. Studies have shown that early in life, before infections become a problem, inflammation is already setting in.
"Babies with cystic fibrosis can seem totally fine with normal respiratory function, but the reality is that the disease is already having an effect," said Emanuela Bruscia, an associate professor of pediatrics at Yale School of Medicine and senior author of the study. "Mucus is already accumulating, and there are areas in the lung with inflammation. And inflammation, if not controlled, is bad for any type of tissue."
In the last decade, new treatments for cystic fibrosis have helped extend life expectancy beyond 50. Those treatments, called CFTR modulators, target the malfunctioning protein—CFTR, or cystic fibrosis transmembrane conductance regulator—that causes the symptoms of cystic fibrosis. But while CFTR modulators help clear mucus in the lungs and maintain lung function, they don't fully address inflammation.
In the new study, the researchers analyzed lung tissue taken from patients with advanced cystic fibrosis to better understand what drives inflammation in the lungs. They found that in the areas of tissue damage, there were a lot of monocytes, up to five to 10 times more than were found in healthy lungs. Then, to evaluate the monocytes' role in cystic fibrosis, they turned to a mouse model of chronic lung inflammation that displays levels of lung damage and functional decline that are similar to what's observed in patients with cystic fibrosis.
"The question was how those cells participate in the disease," said Bruscia.
They found that the monocytes, once drawn into the lungs from the bloodstream, release chemical attractants that pull another type of immune cell called neutrophils into the lung. And the neutrophils cause tissue damage.
This can happen in healthy lungs as well, including as a response to infection. But once the job of the neutrophils and monocytes is done, those cells should leave. In cystic fibrosis, the researchers found, they don't.
"Pro-inflammatory monocytes are part of the normal immune response, but once they arrive and do their job, they should be instructed to leave and be silent," said Bruscia. "But in cystic fibrosis, they arrive, they are super inflammatory, and then they are in an environment in which they can't leave and be silent, so instead, they keep producing these pro-inflammatory mediators."
Because monocytes and neutrophils play a key role in the immune response, getting rid of them altogether would not be beneficial, especially for patients with cystic fibrosis battling chronic lung infections. Instead, Bruscia and her colleagues explored how to reduce the level of monocytes recruited into the lung and bring their number down to levels found in healthy lungs.
In their mouse model, they tested a small molecule called a CCR2 inhibitor. The monocytes found in excess in cystic fibrosis lungs have a protein called C-C chemokine receptor type 2, or CCR2, on their surface. The protein serves as a signal detector. And when an immune signal called a chemokine binds to CCR2, it causes the monocyte to move to where it's needed. By inhibiting CCR2 with the drug, the researchers were able to reduce the number of monocytes recruited into the lungs of mice and slow the progression of tissue damage.
"Importantly, the CCR2 inhibitor didn't block all of the monocytes. It just brought the numbers down closer to healthy levels," said Bruscia.
CCR2 inhibitors are currently undergoing clinical trials for other diseases, like cancer. The findings of this study suggest they could also be effective in treating the chronic inflammation found in cystic fibrosis.
Bruscia and her colleagues are continuing to assess the effectiveness of CCR2 inhibitors in cystic fibrosis models, investigating how they work in the context of lung infection and comparing different types to see which are most effective.
They'll also continue to study what happens at the immunological level in the lungs of patients with cystic fibrosis.
"There's so much going on in the lungs that we don't yet understand," said Bruscia. "This is just the tip of the iceberg."
More information: Hasan H. Öz et al, Recruited monocytes/macrophages drive pulmonary neutrophilic inflammation and irreversible lung tissue remodeling in cystic fibrosis, Cell Reports (2022). DOI: 10.1016/j.celrep.2022.111797