Researchers uncover mechanism behind metabolic vulnerability of some breast cancers

Scientists have known since the 1980s that many cancer cells are relatively sensitive to the deprivation of an essential amino acid known as methionine. It has, however, long been unclear what causes such marked dependency on methionine. Now, a Ludwig Cancer Research study published in the journal Science Signaling and led by Alex Toker, an investigator in the Ludwig Center at Harvard, has elucidated one mechanism behind that dependency. The findings suggest what could be a novel approach to treating a variety of breast tumors.
"Most normal cells generally can survive the acute deprivation of methionine, but some tumor cells are very sensitive to its scarcity," says Toker, an investigator at the Ludwig Center at Harvard Medical School. "In this paper, we show how this methionine dependency is controlled by PIK3CA, a gene that drives cancer when it is mutated in a way that hyperactivates its protein product."
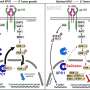
Toker and his colleagues show in their paper that the oncogenic PIK3CA's aberrant activity alters the cell's production of methionine by inhibiting the activity of another protein named xCT, which imports a molecule involved in the cell's production of a related amino acid.
"The concept that all cancers rewire metabolic pathways to fuel the needs of rapidly dividing cells has gained great traction in the cancer community in the last decade or so," Toker said. "We and many other labs have been trying to drill deep in identifying such metabolic vulnerabilities to arrive at novel therapeutic approaches to treating patients."
In their study, Toker's team screened 13 breast cancer lines to identify those that are unable to survive in methionine-starved environments.
When a closer look found that most of these Met-dependent cancer cells also carried mutations in the PIK3CA gene, the scientists hypothesized that the oncogenic PIK3CA might be altering processes involved in methionine metabolism. A review of the published literature revealed that Met-dependency in cancer cells is often correlated with low levels of xCT activity.
This was intriguing because xCT controls the cellular import of cystine (pronounced cys-tine), which is a precursor of the amino acid cysteine (pronounced cys-teen). Like methionine, cysteine is an important building block for proteins. The two amino acids share another similarity: both can be produced from yet another metabolite named homocysteine.
"Homocysteine has two major fates in a given cell," explains study first author Evan Lien, a former graduate student in Toker's lab. "It can either be chemically modified to produce methionine, or it can be shunted towards the synthesis of cysteine."
The scientists speculated that in cancer cells where oncogenic PIK3CA is blocking cystine import, homocysteine would be used to make cysteine. This would leave less homocysteine available to produce methionine, making those breast cancer cells more dependent on an external supply of methionine. To accomplish that, Toker and his team used an existing drug called sulfasalazine that is known to inhibit xCT.
When hit with sulfasalazine, breast cancer cells that would normally survive in methionine-starved environments were no longer able to do so. In effect, the drug made the cells methionine-dependent by mimicking the effects of a mutant PIK3CA gene.
The new findings raise the possibility of killing tumors by triggering methionine dependency using sulfasalazine or other drugs.
"The idea would be to target tumors with a drug that blocks xCT," says Toker. "By doing this, a tumor cell would push homocysteine toward cysteine biosynthesis and the cell would ultimately die due to methionine deprivation, while leaving normal cells unaffected."
Toker and his team are now working to evaluate the feasibility of that approach.
More information: Evan C. Lien et al, Oncogenic PI3K promotes methionine dependency in breast cancer cells through the cystine-glutamate antiporter xCT, Science Signaling (2017). DOI: 10.1126/scisignal.aao6604